Setting up and running absolute binding free energy calculations#
This tutorial gives a step-by-step process to set up an absolute binding free energy (ABFE) simulation campaign using OpenFE. In this tutorial we are performing an absolute binding free energy calculation of toluene binding to T4 Lysozyme.
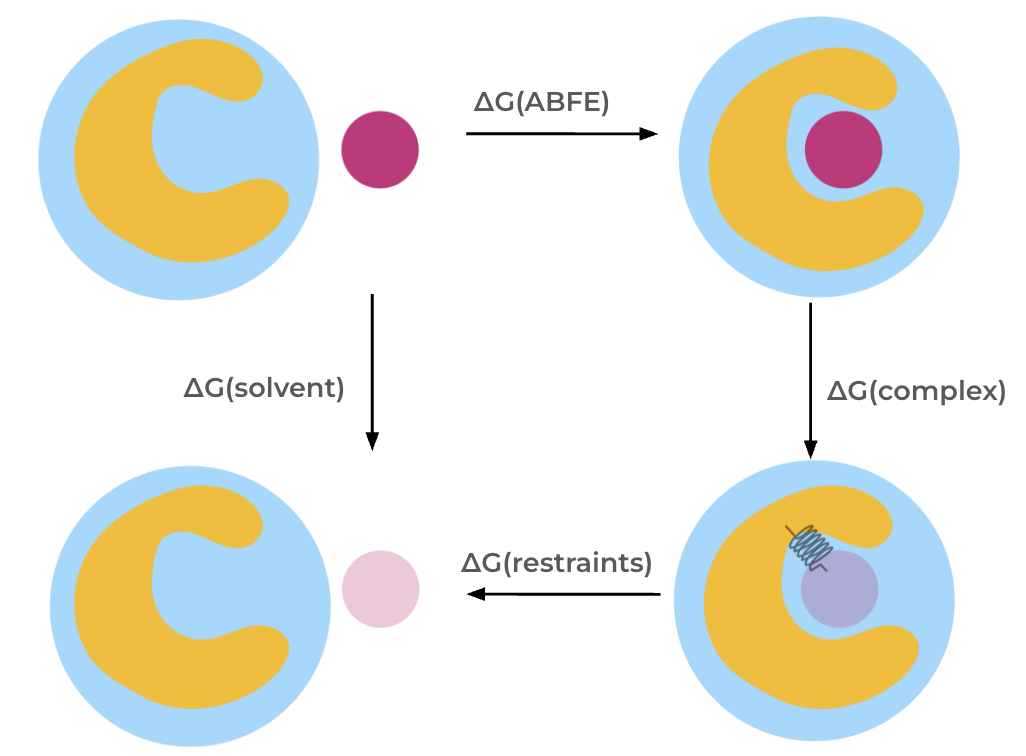
The absolute binding free energy is obtained through a thermodynamic cycle. The interactions of the molecule are decoupled both in solvent, giving \(\Delta G\)(solvent) and in the complex, giving \(\Delta G\)(complex), which allows calculation of the absolute binding free energy, \(\Delta G\)(ABFE).
Note: In this Protocol, the coulombic interactions of the molecule are fully turned off (annihilated), while the Lennard-Jones interactions are decoupled, meaning the intermolecular interactions are turned off, whilst keeping the intramolecular Lennard-Jones interactions.
 #
#
0. Setup for Google Colab#
If you are running this example in Google Colab, run the following cells to setup the environment. If you are running this notebook locally, skip down to 1. Loading the ligand
[ ]:
# NBVAL_SKIP
# Only run this cell if on google colab
import os
if "COLAB_RELEASE_TAG" in os.environ:
# fix for colab's torchvision causing issues
!rm -r /usr/local/lib/python3.12/dist-packages/torchvision
!pip install -q condacolab
import condacolab
condacolab.install_from_url("https://github.com/OpenFreeEnergy/openfe/releases/download/v1.7.0/OpenFEforge-1.7.0-Linux-x86_64.sh")
[ ]:
# NBVAL_SKIP
# Only run this cell if on google colab
import os
if "COLAB_RELEASE_TAG" in os.environ:
import condacolab
import locale
locale.getpreferredencoding = lambda: "UTF-8"
!mkdir inputs && cd inputs && openfe fetch rbfe-tutorial
# quick fix for https://github.com/conda-incubator/condacolab/issues/75
# if deprecation warnings persist, rerun this cell
import warnings
warnings.filterwarnings(action="ignore", message=r"datetime.datetime.utcnow")
for _ in range(3):
# Sometimes we have to re-run the check
try:
condacolab.check()
except:
pass
else:
break
1. Loading the ligand#
First we must load the chemical models for which we wish to calculate the binding free energy. In this example these are initially stored in a molfile (.sdf). This can be loaded using the SDMolSupplier class from rdkit and passed to openfe.
[1]:
%matplotlib inline
import openfe
/home/ialibay/software/mambaforge/install/envs/openfe/lib/python3.12/site-packages/openmoltools/utils.py:9: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import resource_filename
[2]:
from rdkit import Chem
supp = Chem.SDMolSupplier("../cookbook/assets/toluene.sdf", removeHs=False)
ligands = [openfe.SmallMoleculeComponent.from_rdkit(mol) for mol in supp]
2. Charging the ligand#
It is recommended to use a single set of charges for each ligand to ensure reproducibility between repeats.
Here we will use some utility functions from OpenFE which can assign partial charges to a series of molecules with a variety of methods which can be configured via the OpenFFPartialChargeSettings class. In this example we will charge the ligands using the am1bcc method from ambertools which is the default charge scheme used by OpenFE.
[3]:
from openfe.protocols.openmm_utils.omm_settings import OpenFFPartialChargeSettings
from openfe.protocols.openmm_utils.charge_generation import bulk_assign_partial_charges
charge_settings = OpenFFPartialChargeSettings(partial_charge_method="am1bcc", off_toolkit_backend="ambertools")
ligands = bulk_assign_partial_charges(
molecules=ligands,
overwrite=False,
method=charge_settings.partial_charge_method,
toolkit_backend=charge_settings.off_toolkit_backend,
generate_n_conformers=charge_settings.number_of_conformers,
nagl_model=charge_settings.nagl_model,
processors=1
)
Generating charges: 100%|█████████████████████████| 1/1 [00:00<00:00, 1.44it/s]
3. Creating ChemicalSystems#
OpenFE describes complex molecular systems as being composed of Components. For example, we have a SmallMoleculeComponent for each small molecule, a SolventComponent to describe the solvent, and a ProteinComponent to describe the protein.
The Components are joined in a ChemicalSystem to define the entire system.
In state A of the binding free energy Protocol, the ligand is fully interacting in the complex and the ChemicalSystem contains the ligand, the protein, and the solvent. In the other endstate, state B, the ligand is fully decoupled in the complex. Therefore, the ChemicalSystem in state B only contains the protein and the solvent.
Note that for ABFE simulations, we are not separately defining the solvent state, but the Protocol creates that based on the complex states.
[4]:
# defaults are water with NaCl at 0.15 M
solvent = openfe.SolventComponent()
[5]:
protein = openfe.ProteinComponent.from_pdb_file("../cookbook/assets/t4_lysozyme.pdb")
[6]:
# In state A the ligand is fully interacting in the solvent
systemA = openfe.ChemicalSystem({
'ligand': ligands[0],
'protein': protein,
'solvent': solvent,
}, name=ligands[0].name)
# In state B the ligand is fully decoupled in the solvent, therefore we are only defining the solvent here
systemB = openfe.ChemicalSystem({
'protein': protein,
'solvent': solvent,
})
4. Defining the ABFE simulation settings and creating a Protocol#
There are various different parameters which can be set to determine how the ABFE simulation will take place.
The easiest way to customize Protocol settings is to start with the default settings, and modify them. Many settings carry units with them.
[7]:
from openfe.protocols.openmm_afe import AbsoluteBindingProtocol;
/home/ialibay/software/mambaforge/install/envs/openfe/lib/python3.12/site-packages/Bio/Application/__init__.py:39: BiopythonDeprecationWarning: The Bio.Application modules and modules relying on it have been deprecated.
Due to the on going maintenance burden of keeping command line application
wrappers up to date, we have decided to deprecate and eventually remove these
modules.
We instead now recommend building your command line and invoking it directly
with the subprocess module.
warnings.warn(
[8]:
settings = AbsoluteBindingProtocol.default_settings()
[9]:
settings
{'alchemical_settings': {},
'complex_equil_output_settings': {'checkpoint_interval': {'unit': 'nanosecond',
'val': 1.0},
'checkpoint_storage_filename': 'checkpoint.chk',
'equil_npt_structure': 'equil_npt_structure.pdb',
'equil_nvt_structure': 'equil_nvt_structure.pdb',
'forcefield_cache': 'db.json',
'log_output': 'production_equil_simulation.log',
'minimized_structure': 'minimized.pdb',
'output_indices': 'all',
'preminimized_structure': 'system.pdb',
'production_trajectory_filename': 'production_equil.xtc',
'trajectory_write_interval': {'unit': 'picosecond',
'val': 20.0}},
'complex_equil_simulation_settings': {'equilibration_length': {'unit': 'nanosecond',
'val': 0.5},
'equilibration_length_nvt': {'unit': 'nanosecond',
'val': 0.25},
'minimization_steps': 5000,
'production_length': {'unit': 'nanosecond',
'val': 5.0}},
'complex_lambda_settings': {'lambda_elec': [0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.1,
0.2,
0.3,
0.4,
0.5,
0.6,
0.7,
0.8,
0.9,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0],
'lambda_restraints': [0.0,
0.2,
0.4,
0.6,
0.8,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0],
'lambda_vdw': [0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.1,
0.2,
0.3,
0.4,
0.5,
0.6,
0.65,
0.7,
0.75,
0.8,
0.85,
0.9,
0.95,
1.0]},
'complex_output_settings': {'checkpoint_interval': {'unit': 'nanosecond',
'val': 1.0},
'checkpoint_storage_filename': 'complex_checkpoint.nc',
'forcefield_cache': 'db.json',
'output_filename': 'complex.nc',
'output_indices': 'not water',
'output_structure': 'alchemical_system.pdb',
'positions_write_frequency': {'unit': 'picosecond',
'val': 100.0},
'velocities_write_frequency': None},
'complex_simulation_settings': {'early_termination_target_error': {'unit': 'kilocalorie_per_mole',
'val': 0.0},
'equilibration_length': {'unit': 'nanosecond',
'val': 1},
'minimization_steps': 5000,
'n_replicas': 30,
'production_length': {'unit': 'nanosecond',
'val': 10.0},
'real_time_analysis_interval': {'unit': 'picosecond',
'val': 250.0},
'real_time_analysis_minimum_time': {'unit': 'picosecond',
'val': 500.0},
'sampler_method': 'repex',
'sams_flatness_criteria': 'logZ-flatness',
'sams_gamma0': 1.0,
'time_per_iteration': {'unit': 'picosecond',
'val': 2.5}},
'complex_solvation_settings': {'box_shape': 'dodecahedron',
'box_size': None,
'box_vectors': None,
'number_of_solvent_molecules': None,
'solvent_model': 'tip3p',
'solvent_padding': {'unit': 'nanometer',
'val': 1.0}},
'engine_settings': {'compute_platform': 'cuda', 'gpu_device_index': None},
'forcefield_settings': {'constraints': 'hbonds',
'forcefields': ['amber/ff14SB.xml',
'amber/tip3p_standard.xml',
'amber/tip3p_HFE_multivalent.xml',
'amber/phosaa10.xml'],
'hydrogen_mass': 3.0,
'nonbonded_cutoff': {'unit': 'nanometer', 'val': 0.9},
'nonbonded_method': 'PME',
'rigid_water': True,
'small_molecule_forcefield': 'openff-2.2.1'},
'integrator_settings': {'barostat_frequency': {'unit': 'timestep',
'val': 25.0},
'constraint_tolerance': 1e-06,
'langevin_collision_rate': {'unit': '1 / picosecond',
'val': 1.0},
'n_restart_attempts': 20,
'reassign_velocities': False,
'remove_com': False,
'timestep': {'unit': 'femtosecond', 'val': 4.0}},
'partial_charge_settings': {'nagl_model': None,
'number_of_conformers': None,
'off_toolkit_backend': 'ambertools',
'partial_charge_method': 'am1bcc'},
'protocol_repeats': 3,
'restraint_settings': {'K_phiA': {'unit': 'kilojoule_per_mole / radian ** 2',
'val': 334.72},
'K_phiB': {'unit': 'kilojoule_per_mole / radian ** 2',
'val': 334.72},
'K_phiC': {'unit': 'kilojoule_per_mole / radian ** 2',
'val': 334.72},
'K_r': {'unit': 'kilojoule_per_mole / nanometer ** 2',
'val': 4184.0},
'K_thetaA': {'unit': 'kilojoule_per_mole / radian ** 2',
'val': 334.72},
'K_thetaB': {'unit': 'kilojoule_per_mole / radian ** 2',
'val': 334.72},
'anchor_finding_strategy': 'bonded',
'dssp_filter': True,
'host_max_distance': {'unit': 'nanometer', 'val': 1.5},
'host_min_distance': {'unit': 'nanometer', 'val': 0.5},
'host_selection': 'backbone',
'rmsf_cutoff': {'unit': 'nanometer', 'val': 0.1}},
'solvent_equil_output_settings': {'checkpoint_interval': {'unit': 'nanosecond',
'val': 1.0},
'checkpoint_storage_filename': 'checkpoint.chk',
'equil_npt_structure': 'equil_npt_structure.pdb',
'equil_nvt_structure': 'equil_nvt_structure.pdb',
'forcefield_cache': 'db.json',
'log_output': 'production_equil_simulation.log',
'minimized_structure': 'minimized.pdb',
'output_indices': 'all',
'preminimized_structure': 'system.pdb',
'production_trajectory_filename': 'production_equil.xtc',
'trajectory_write_interval': {'unit': 'picosecond',
'val': 20.0}},
'solvent_equil_simulation_settings': {'equilibration_length': {'unit': 'nanosecond',
'val': 0.2},
'equilibration_length_nvt': {'unit': 'nanosecond',
'val': 0.1},
'minimization_steps': 5000,
'production_length': {'unit': 'nanosecond',
'val': 0.5}},
'solvent_lambda_settings': {'lambda_elec': [0.0,
0.25,
0.5,
0.75,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0,
1.0],
'lambda_restraints': [0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0,
0.0],
'lambda_vdw': [0.0,
0.0,
0.0,
0.0,
0.0,
0.12,
0.24,
0.36,
0.48,
0.6,
0.7,
0.77,
0.85,
1.0]},
'solvent_output_settings': {'checkpoint_interval': {'unit': 'nanosecond',
'val': 1.0},
'checkpoint_storage_filename': 'solvent_checkpoint.nc',
'forcefield_cache': 'db.json',
'output_filename': 'solvent.nc',
'output_indices': 'not water',
'output_structure': 'alchemical_system.pdb',
'positions_write_frequency': {'unit': 'picosecond',
'val': 100.0},
'velocities_write_frequency': None},
'solvent_simulation_settings': {'early_termination_target_error': {'unit': 'kilocalorie_per_mole',
'val': 0.0},
'equilibration_length': {'unit': 'nanosecond',
'val': 1.0},
'minimization_steps': 5000,
'n_replicas': 14,
'production_length': {'unit': 'nanosecond',
'val': 10.0},
'real_time_analysis_interval': {'unit': 'picosecond',
'val': 250.0},
'real_time_analysis_minimum_time': {'unit': 'picosecond',
'val': 500.0},
'sampler_method': 'repex',
'sams_flatness_criteria': 'logZ-flatness',
'sams_gamma0': 1.0,
'time_per_iteration': {'unit': 'picosecond',
'val': 2.5}},
'solvent_solvation_settings': {'box_shape': 'dodecahedron',
'box_size': None,
'box_vectors': None,
'number_of_solvent_molecules': None,
'solvent_model': 'tip3p',
'solvent_padding': {'unit': 'nanometer',
'val': 1.5}},
'thermo_settings': {'ph': None,
'pressure': {'unit': 'bar', 'val': 1},
'redox_potential': None,
'temperature': {'unit': 'kelvin', 'val': 298.15}}}
/home/ialibay/software/mambaforge/install/envs/openfe/lib/python3.12/site-packages/gufe/settings/models.py:30: PydanticDeprecatedSince20: The `dict` method is deprecated; use `model_dump` instead. Deprecated in Pydantic V2.0 to be removed in V3.0. See Pydantic V2 Migration Guide at https://errors.pydantic.dev/2.11/migration/
pprint.pprint(self.dict())
By default we run 3 repeats with solvent and complex simulation lengths of 10 ns over 14 and 30 lambda windows. To speed things up here we instead run 1 repeat with both solvent and complex simulation lengths of 0.5 ns over 14 and 30 lambda windows.
Changing default values:
[10]:
from openff.units import unit
# Run only a single repeat
settings.protocol_repeats = 1
# Change the min and max distance between protein and ligand atoms for Boresch restraints to avoid periodicity issues
settings.restraint_settings.host_min_distance = 0.5 * unit.nanometer
settings.restraint_settings.host_max_distance = 1.5 * unit.nanometer
# The compute platform is CUDA by default, but we're just showing you how you could change it
settings.engine_settings.compute_platform = 'CUDA'
Creating the Protocol#
With the Settings inspected and adjusted, we can provide these to the Protocol. This Protocol defines the procedure to estimate a free energy difference between two chemical systems, with the details of the two end states yet to be defined.
[11]:
protocol = AbsoluteBindingProtocol(settings=settings)
5. Running the ABFE simulation#
(a) Using the CLI#
Once we have the ChemicalSystems, and the Protocol, we can create the Transformation.
[12]:
transformation = openfe.Transformation(
stateA=systemA,
stateB=systemB,
mapping=None,
protocol=protocol, # use protocol created above
name=f"{systemA.name}"
)
We’ll write out the transformation to disk, so that it can be run using the openfe quickrun command:
[13]:
import pathlib
# first we create the directory
transformation_dir = pathlib.Path("abfe_json")
transformation_dir.mkdir(exist_ok=True)
# then we write out the transformation
transformation.dump(transformation_dir / f"{transformation.name}.json")
/home/ialibay/software/mambaforge/install/envs/openfe/lib/python3.12/site-packages/gufe/transformations/transformation.py:124: DeprecationWarning: use of this method is deprecated; instead use `to_json`
warnings.warn(
You can run the ABFE simulation from the CLI by using the openfe quickrun command. It takes a transformation JSON as input, and the flags -o to give the final output JSON file and -d for the directory where simulation results should be stored. For example,
openfe quickrun path/to/transformation.json -o results.json -d working-directory
where path/to/transformation.json is the path to one of the files created above.
(b) Using the Python API#
Creating the ``ProtocolDAG``
Once we have the two ChemicalSystems, and the Protocol, we can create the ProtocolDAG.
This creates a directed-acyclic-graph (DAG) of computational tasks necessary for creating an estimate of the free energy difference between the two chemical systems.
[14]:
dag = protocol.create(stateA=systemA, stateB=systemB, mapping=None)
To summarize, this ProtocolDAG contains:
chemical models of both sides of the alchemical transformation in
systemAandsystemBa description of the exact computational algorithm to use to perform the estimate in
Protocolthe
mappingis set toNonesince no atoms are mapped in the ABFE protocol
Executing the simulation
The DAG contains many invdividual jobs. We could execute them sequentially in this notebook using the gufe.protocols.execute function, however, this calculation is too expensive to be run in that manner, therefore the execulte_DAG command is commented out here.
In a more realistic (expansive) situation we would farm off the individual jobs to a HPC cluster or cloud compute service so they could be executed in parallel.
Note: we use the shared_basedir and scratch_basedir argument of execute_DAG in order to set the directory where the simulation files are written to
[15]:
from gufe.protocols import execute_DAG
import pathlib
[16]:
# Finally we can run the simulations
path = pathlib.Path('./abfe_results')
# Commented out since this would be too expensive to run in this notebook.
# path.mkdir()
# Execute the DAG
# dag_results = execute_DAG(dag, scratch_basedir=path, shared_basedir=path, n_retries=3)
6. Analysis#
Finally now that we’ve run our simulations, let’s go ahead and gather the free energies for both phases.
Python API#
If you executed the simulations using the Python API, you will have generated a dag_results object. You can analyze these results by calling the Protocols’ gather() method. This takes a list of completed DAG results and returns a AbsoluteBindingProtocolResult which can return a free energy estimate and uncertainty by calling the get_estimate() and get_uncertainty() methods.
The code for this part is commented out since executing the DAG in this notebook would be too expensive.
[17]:
# Get the complex and solvent results
# protocol_results = protocol.gather([dag_results])
# print(f"ABFE dG: {protocol_results.get_estimate()}, err {protocol_results.get_uncertainty()}")
You can save the AbsoluteBindingProtocolResult to a JSON output file in the following manner:
[18]:
# Save the results in a json file
# import gzip
# import json
# import gufe
# outdict = {
# "estimate": protocol_results.get_estimate(),
# "uncertainty": protocol_results.get_uncertainty(),
# "protocol_result": protocol_results.to_dict(),
# "unit_results": {
# unit.key: unit.to_keyed_dict()
# for unit in dag_results.protocol_unit_results
# }
# }
# with open("abfe_json/toluene_results.json") as stream:
# json.dump(outdict, stream, cls=gufe.tokenization.JSON_HANDLER.encoder)
CLI / Quickrun#
If you ran the simulation using the CLI (i.e. by calling openfe quickrun ) you will end up with the same JSON output file as the one created in the previous cell. To get your simulation results you can load them back into Python in the following manner:
[19]:
import gzip
import json
import gufe
outfile = "abfe_results/toluene_results.json"
with open(outfile) as stream:
results = json.load(stream)
estimate = results['estimate']
uncertainty = results['uncertainty']
[20]:
estimate
[20]:
{'magnitude': -4.671169105743085,
'unit': 'kilocalorie_per_mole',
':is_custom:': True,
'pint_unit_registry': 'openff_units'}