Running a Molecular Dynamics (MD) simulation of a protein-ligand complex#
In this notebook we run an MD simulation of benzene bound to T4-lysozyme L99A.
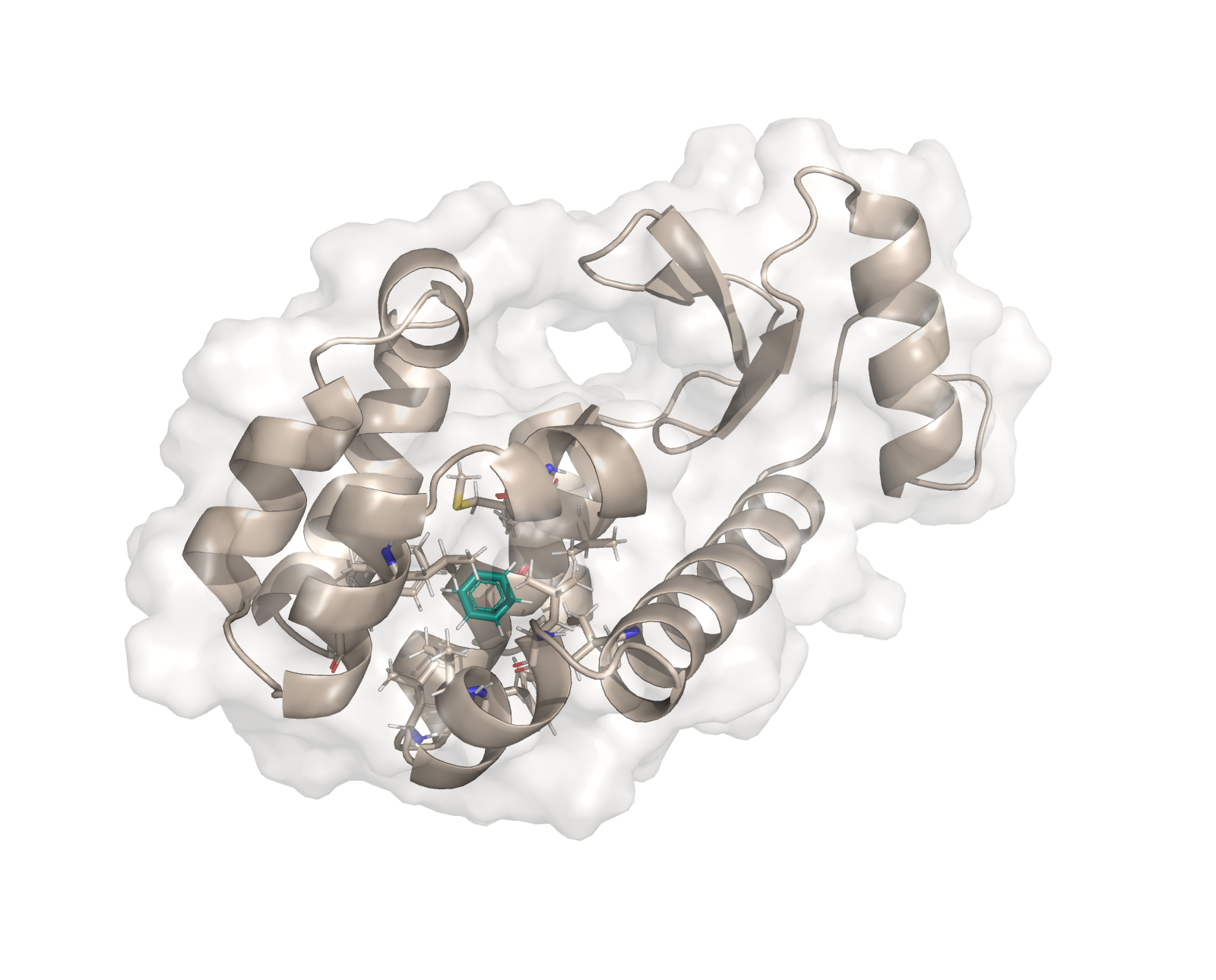
The plain MD protocol allows the user to run an MD simulation either in solvent or vacuum of e.g. a small molecule, a protein, or a protein-ligand complex. Running an MD simulations can be useful for a variety of things, such as pre-equilibration of a protein-ligand complex prior to running free energy calculations or for getting insights into the general dynamics of the system of interest.
The MD protocol uses different software tools in a series of steps and provides multiple outputs for the user:
Step |
Software used |
Outputs (with default names) |
|---|---|---|
|
OpenFE, Gufe, RDKit |
|
|
OpenFE - OpenMMForceFields - OpenFF |
Forcefield cache ( |
|
OpenFE - OpenMM + OpenMMTools |
Structure of the full system ( |
|
OpenFE - OpenMM + OpenMMTools |
Minimized Structure ( |
|
OpenFE - OpenMM + OpenMMTools |
NVT equilibrated structure ( |
|
OpenFE - OpenMM + OpenMMTools |
NPT equilibrated structure ( |
|
OpenFE - OpenMM + OpenMMTools |
Simulation trajectory ( |
0. Setup for Google Colab#
If you are running this example in Google Colab, run the following cells to setup the environment. If you are running this notebook locally, skip down to 1. Overview
[ ]:
# NBVAL_SKIP
# Only run this cell if on google colab
import os
if "COLAB_RELEASE_TAG" in os.environ:
# fix for colab's torchvision causing issues
!rm -r /usr/local/lib/python3.12/dist-packages/torchvision
!pip install -q condacolab
import condacolab
condacolab.install_from_url("https://github.com/OpenFreeEnergy/openfe/releases/download/v1.7.0/OpenFEforge-1.7.0-Linux-x86_64.sh")
[ ]:
# NBVAL_SKIP
# Only run this cell if on google colab
import os
if "COLAB_RELEASE_TAG" in os.environ:
import condacolab
import locale
locale.getpreferredencoding = lambda: "UTF-8"
!mkdir inputs && cd inputs && openfe fetch rbfe-tutorial
# quick fix for https://github.com/conda-incubator/condacolab/issues/75
# if deprecation warnings persist, rerun this cell
import warnings
warnings.filterwarnings(action="ignore", message=r"datetime.datetime.utcnow")
for _ in range(3):
# Sometimes we have to re-run the check
try:
condacolab.check()
except:
pass
else:
break
1. Defining the ChemicalSystem#
ChemicalSystems are OpenFE containers which define the various components which exist in a system of interest. Here, we will be passing the SmallMoleculeComponent for benzene, a ProteinComponent generated from a PDB file, and a SolventComponent which will contain the necessary information for OpenMM’s Modeller class to add water and 0.15 M NaCl around the solute.
[3]:
import openfe
from openfe import ChemicalSystem, ProteinComponent, SmallMoleculeComponent, SolventComponent
from openff.units import unit
# Define the ligand we are interested in
ligand = SmallMoleculeComponent.from_sdf_file('assets/benzene.sdf')
# Define the solvent environment and protein structure
solvent = SolventComponent(ion_concentration=0.15 * unit.molar)
protein = ProteinComponent.from_pdb_file('assets/t4_lysozyme.pdb', name='t4-lysozyme')
# create the ChemicalSystem
system = ChemicalSystem({'ligand': ligand, 'protein': protein, 'solvent': solvent}, name=f"{ligand.name}_{protein.name}")
2. Defining the MD simulation settings#
There are various different parameters which can be set to determine how the MD simulation will take place. To allow for maximum user flexibility, these are defined as a series of settings objects which control the following:
Setting |
Description |
|---|---|
|
Parameters controlling the simulation plan, including the number of |
|
Parameters controlling the output from the MD simulations, including file names to save the system after minimization, NVT and NPT equilibration, and production run. Special |
|
Settings that define the forcefield for the components, including the general |
|
Parameters determining how the OpenMM engine will execute the simulation. This controls the |
|
Parameters controlling the LangevinSplittingDynamicsMove integrator used for simulation, as well as the |
|
Settings that define which method is used for assigning partial charges. |
|
Defines how often to run the MD protocol. |
|
Parameters to control the |
|
Parameters to control e.g. the |
The easiest way to access and change settings is by first importing the default settings, printing them and then changing the settings according to the user’s needs.
[4]:
from openfe.protocols.openmm_md.plain_md_methods import PlainMDProtocol
from openff.units import unit
settings = PlainMDProtocol.default_settings()
settings.simulation_settings.equilibration_length_nvt = 0.01 * unit.nanosecond # setting the nvt equilibration length to 10 ps
settings.simulation_settings.equilibration_length = 0.01 * unit.nanosecond # setting the npt equilibration length to 10 ps
# Setting the production length and checkpoint interval to 20 ps to match the trajectory write interval, so one frame will be written
settings.simulation_settings.production_length = 0.02 * unit.nanosecond # setting the npt production length to 20 ps
settings.output_settings.checkpoint_interval = 0.02 * unit.nanosecond # setting the checkpoint interval to 20 ps
settings.engine_settings.compute_platform = 'CPU' # running the simulation on the cpu
settings.solvation_settings.solvent_padding = 1.0 * unit.nanometer # set the solvent padding to 1 nm to reduce the number of waters
[5]:
settings
{'engine_settings': {'compute_platform': 'CPU', 'gpu_device_index': None},
'forcefield_settings': {'constraints': 'hbonds',
'forcefields': ['amber/ff14SB.xml',
'amber/tip3p_standard.xml',
'amber/tip3p_HFE_multivalent.xml',
'amber/phosaa10.xml'],
'hydrogen_mass': 3.0,
'nonbonded_cutoff': {'unit': 'nanometer', 'val': 0.9},
'nonbonded_method': 'PME',
'rigid_water': True,
'small_molecule_forcefield': 'openff-2.2.1'},
'integrator_settings': {'barostat_frequency': {'unit': 'timestep',
'val': 25.0},
'constraint_tolerance': 1e-06,
'langevin_collision_rate': {'unit': '1 / picosecond',
'val': 1.0},
'n_restart_attempts': 20,
'reassign_velocities': False,
'remove_com': False,
'timestep': {'unit': 'femtosecond', 'val': 4.0}},
'output_settings': {'checkpoint_interval': {'unit': 'nanosecond', 'val': 0.02},
'checkpoint_storage_filename': 'checkpoint.chk',
'equil_npt_structure': 'equil_npt.pdb',
'equil_nvt_structure': 'equil_nvt.pdb',
'forcefield_cache': 'db.json',
'log_output': 'simulation.log',
'minimized_structure': 'minimized.pdb',
'output_indices': 'not water',
'preminimized_structure': 'system.pdb',
'production_trajectory_filename': 'simulation.xtc',
'trajectory_write_interval': {'unit': 'picosecond',
'val': 20.0}},
'partial_charge_settings': {'nagl_model': None,
'number_of_conformers': None,
'off_toolkit_backend': 'ambertools',
'partial_charge_method': 'am1bcc'},
'protocol_repeats': 1,
'simulation_settings': {'equilibration_length': {'unit': 'nanosecond',
'val': 0.01},
'equilibration_length_nvt': {'unit': 'nanosecond',
'val': 0.01},
'minimization_steps': 5000,
'production_length': {'unit': 'nanosecond',
'val': 0.02}},
'solvation_settings': {'box_shape': 'dodecahedron',
'box_size': None,
'box_vectors': None,
'number_of_solvent_molecules': None,
'solvent_model': 'tip3p',
'solvent_padding': {'unit': 'nanometer', 'val': 1.0}},
'thermo_settings': {'ph': None,
'pressure': {'unit': 'bar', 'val': 1},
'redox_potential': None,
'temperature': {'unit': 'kelvin', 'val': 298.15}}}
/Users/atravitz/micromamba/envs/openfe-conda/lib/python3.11/site-packages/gufe/settings/models.py:30: PydanticDeprecatedSince20: The `dict` method is deprecated; use `model_dump` instead. Deprecated in Pydantic V2.0 to be removed in V3.0. See Pydantic V2 Migration Guide at https://errors.pydantic.dev/2.11/migration/
pprint.pprint(self.dict())
3. Creating a Protocol#
The actual simulation is performed by a `Protocol <https://docs.openfree.energy/en/stable/guide/models/execution.html#protocols-and-the-execution-model>`__.
With the Settings inspected and adjusted, we can provide these to the Protocol. Here, the OpenMM-based MD Protocol is named PlainMDProtocol.
[6]:
# Creating the Protocol
from openfe.protocols.openmm_md.plain_md_methods import PlainMDProtocol
protocol = PlainMDProtocol(settings=settings)
4. Creating the NonTransformation#
Once we have the ChemicalSystems, and the Protocol, we can create the NonTransformation. NonTransformation here simply means that the system is not “transformed” between two end states as is the case in binding free energy calculations.
[7]:
nontransformation = openfe.NonTransformation(
system=system,
protocol=protocol, # use protocol created above
name=f"{system.name}",
)
5. Running the MD simulation#
There are two ways in which you could execute this MD simulation, either using the OpenFE command-line interface (CLI) or the Python API.
(a) Using the CLI
We’ll write out the transformation to disk, so that it can be run using the openfe quickrun command:
[8]:
import pathlib
# first we create the directory
md_dir = pathlib.Path("md_input")
md_dir.mkdir(exist_ok=True)
# then we write out the transformation
nontransformation.to_json(md_dir / f"{nontransformation.name}.json")
You can run the MD simulation from the CLI by using the openfe quickrun command. It takes a transformation JSON as input, and the flags -o to give the final output JSON file and -d for the directory where simulation results should be stored. For example,
openfe quickrun path/to/nontransformation.json -o results.json -d working-directory
where path/to/nontransformation.json is the path to one of the files created above (md_input/benzene_t4-lysozyme.json).
(b) Using the Python API
Alternatively, the MD simulation can be run by executing the ProtocolDAG. The ProtocolDAG is created using the protocol.create() method and requires as input the ChemicalSystem.
Note: we use the shared_basedir and scratch_basedir argument of execute_DAG in order to set the directory where the simulation files are written to.
[9]:
import gufe
import pathlib
# Creating the Protocol
protocol = PlainMDProtocol(settings=settings)
# Creating the Protocol DAG
dag = protocol.create(stateA=system, stateB=system, mapping=None)
workdir = pathlib.Path('./')
# Running the MD simulations
dagres = gufe.protocols.execute_DAG(
dag,
shared_basedir=workdir,
scratch_basedir=workdir,
keep_shared=True, # set this to True to save the outputs
n_retries=3
)
INFO:openfe.utils.system_probe.log:SYSTEM CONFIG DETAILS:
INFO:openfe.utils.system_probe.log.hostname:hostname: 'Alyssas-Macbook-Pro.local'
INFO:openfe.utils.system_probe.log.gpu:CUDA-based GPU not found
INFO:openfe.utils.system_probe.log:Memory used: 20.7G (63.6%)
INFO:openfe.utils.system_probe.log:scratch_PlainMDProtocolUnit-52e66ff618014bb3877c15dc0d137171_attempt_0: 43% full (530.3G free)
INFO:gufekey.openfe.protocols.openmm_md.plain_md_methods.PlainMDProtocolUnit:Creating system
INFO:openmmforcefields.generators.template_generators:Requested to generate parameters for residue <Residue 0 (UNK) of chain 0>
INFO:openmmforcefields.generators.template_generators:Generating a residue template for [H][c]1[c]([H])[c]([H])[c]([H])[c]([H])[c]1[H] using openff-2.2.1
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 0
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 1
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 2
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 3
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 4
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 5
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 6
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 7
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 8
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 9
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 10
INFO:openff.interchange.smirnoff._nonbonded:Preset charges applied to atom index 11
/Users/atravitz/micromamba/envs/openfe-conda/lib/python3.11/site-packages/openfe/protocols/openmm_utils/omm_compute.py:76: UserWarning: Non-CUDA platform selected: CPU, this may significantly impact simulation performance
warnings.warn(wmsg)
WARNING:root:Non-CUDA platform selected: CPU, this may significantly impact simulation performance
INFO:openfe.protocols.openmm_md.plain_md_methods:minimizing systems
INFO:openfe.protocols.openmm_md.plain_md_methods:Running NVT equilibration
INFO:openfe.protocols.openmm_md.plain_md_methods:Completed NVT equilibration in 19.77936577796936 seconds
INFO:openfe.protocols.openmm_md.plain_md_methods:Running NPT equilibration
INFO:openfe.protocols.openmm_md.plain_md_methods:Completed NPT equilibration in 22.49833083152771 seconds
INFO:openfe.protocols.openmm_md.plain_md_methods:running production phase
INFO:openfe.protocols.openmm_md.plain_md_methods:Completed simulation in 45.916950941085815 seconds
Following files were created for the MD run. Note that ff multiple repeats of the MD simulation were run (protocol_repeats > 1), you will get N full replicates of these output files with N being the number of repeats.
[10]:
!ls shared_PlainMDProtocolUnit-6b85013e6cb94120baf447540650643b_attempt_0/
ls: shared_PlainMDProtocolUnit-6b85013e6cb94120baf447540650643b_attempt_0/: No such file or directory
Performance consideration for gas phase MD simulations#
For gas phase MD simulations, we suggest setting OPENMM_CPU_THREADS to 1 to obtain good performance.
[ ]: