Simulation Setup#
This section provides details on how to set up a free energy calculation or MD simulations.
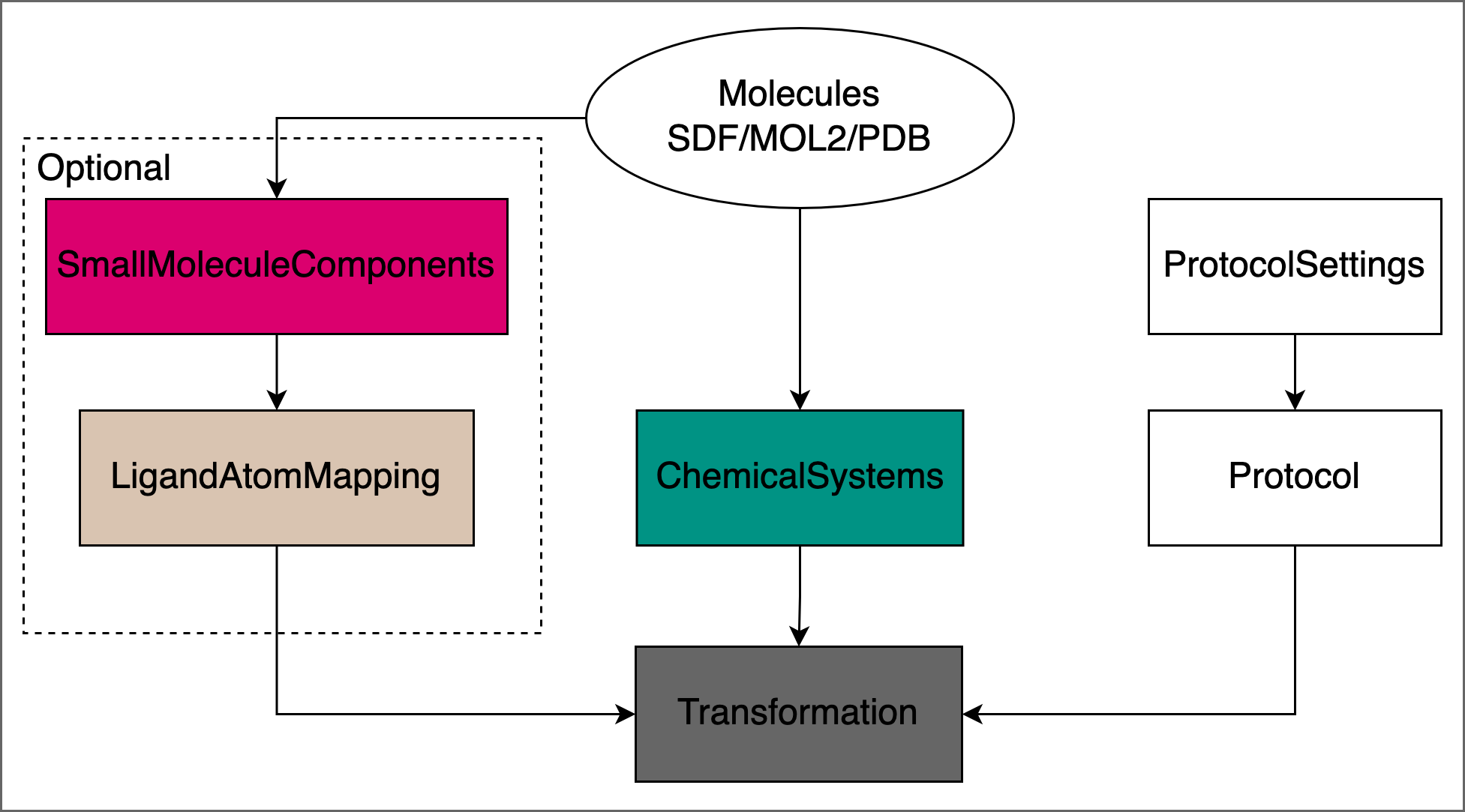
All protocols in OpenFE follow the same general structure:
Reading in input structures and creating
ChemicalSystemsDefining the
Protocolwith specific ProtocolSettings.Creating
LigandAtomMappings for relative free energy calculation Protocols.
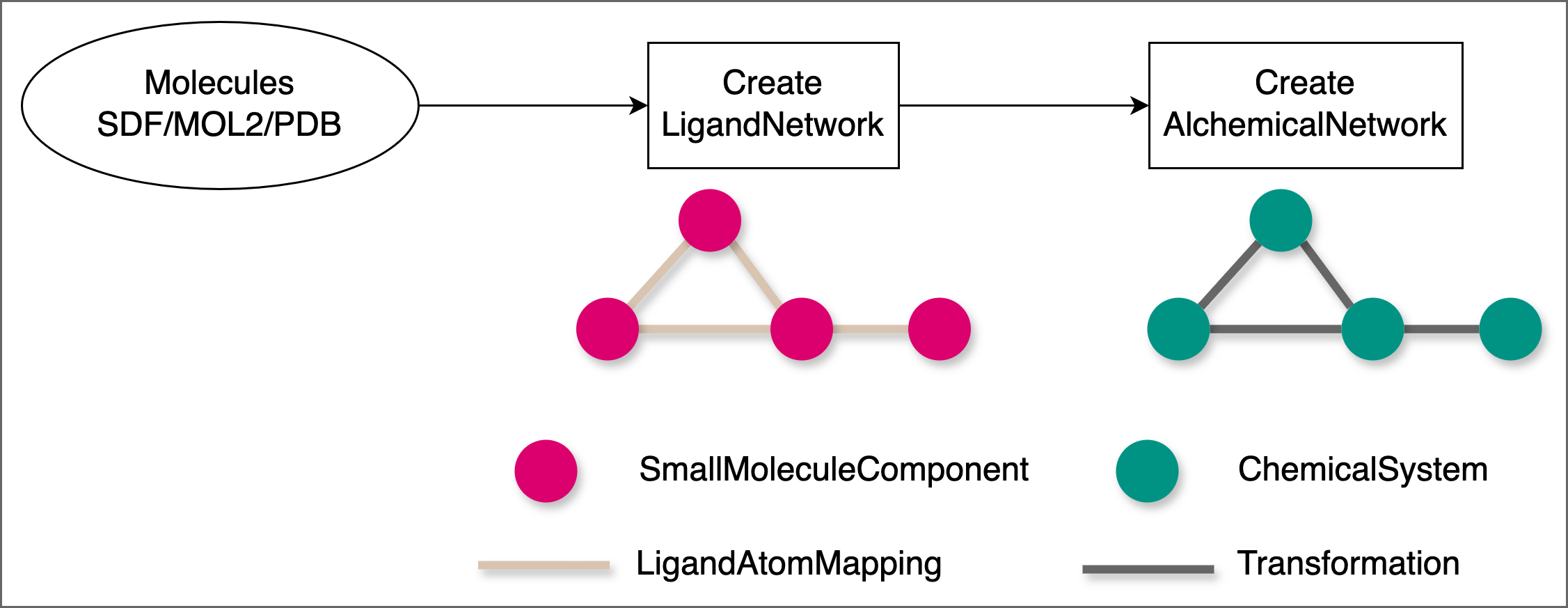
The image below demonstrates how, for relative free energy calculations, you plan a network of ligand transformations starting from input SDF / MOL2 / PDB files:
The procedure for setting up a simulation depends on the type of free energy calculation you are running. More detailed instructions can be found in the following sections:
To set up your alchemical network using the Python interface, but run it using the CLI, you will need to export the network in the same format used by the CLI. See dumping transformations for more details.